
Le varianti virali sono un argomento, ahinoi, attuale ma i cui termini descrittivi sono usati in modo incongruo o non consapevole perfino da alcuni addetti ai lavori, figuriamoci dai media e dalle persone che da questi traggono le informazioni.
Per un riassunto delle varianti di SARS-CoV-2 più rilevanti vi rimando al precedente articolo.
Il tema non è semplice specie quando si parla di microorganismi, non solo per le micro-dimensioni ma per il venir meno di alcune possibilità di categorizzazione basate sulla riproduzione sessuata (specie) che permette di alzare confini, funzionali e non solo morfologici, tra organismi.
Del resto se in ambito animale (vegetale) è l'esame di zoologia I (botanica generale) a fornire le basi per comprendere il grado di parentela facendo riferimento a chiavi di anatomia comparata così da distinguere i casi di evoluzione convergente da quella di "cugini diversi", in ambito microbico l'approccio morfologico serve come prima scrematura e per tutto il resto si deve procede per via genetica.
Esempio banale? Un verme segmentato e un millepiedi appaiono ben più simili che una medusa e un corallo eppure i primi due appartengono a Phyla diversi (anellidi e artropodi, rispettivamente) mentre gli ultimi due sono entrambi cnidari.
Se vogliamo contestualizzare i termini in voga perfino nel linguaggio corrente quando si parla di batteri e virus (ceppo, variante, linea, mutante ...) dobbiamo prenderla un poco alla lontana risalendo alle prime tecniche di classificazione basate su approcci opposti come "raggruppare" o "trovare differenze". Il tutto per risalire a domande come "è una nuova specie?" o "è un nuovo virus?".
Parte fondante del problema è che, per ragioni legate all'evoluzione, tutti gli organismi sono collegati. Differiscono solo per una questione di grado, funzione questa di quanto indietro nel tempo bisogna risalire per trovare l'antenato comune; possiamo visualizzare il concetto pensando al nodo che separa due rami dell’albero (evolutivo).
I tentativi di catalogare la natura sono vecchi quanto (e anche più) Aristotele e hanno visto la lotta tra lumpers (approccio raggruppatore) e splitters (approccio differenziante) con i primi che facevano piu attenzione ai tratti che univano le famiglie e i secondi alle differenze che permettevano di creare nuove famiglie.
A complicare le cose l’assenza di accordo sull'importanza relativa delle caratteristiche misurate; assenza del tutto comprensibile nel mondo pre-molecolare e soprattutto pre-genomico.
A vederla con occhi moderni questa diatriba ha un forte sentore di filosofia e non a torto.
Lo stesso Charles Darwin era ben consapevole del problema e con il suo lavoro pose le basi per una nuova catalogazione, evolutiva oltre che descrittiva.
Da allora il confine tra specie fu tracciato nella compatibilità riproduttiva totale, alias generare progenie ugualmente vitale ma soprattutto fertile. Ecco perché cavalli e asini appartengono a specie diverse (sebbene simili) mentre un boxer e uno yorkshire sono la stessa specie (cani) anche se esteriormente molto diverse. Un punto che indica quanto differenze o somiglianze possano essere fuorvianti (vedi anche nota a fondo pagina su Homo sapiens e neanderthalensis).
Quando si passa a batteri e più ancora ai virus, il primo problema che incontriamo è che stiamo osservando minuscole forme al microscopio di cui possiamo certo distinguere differenze morfologiche come il filamentoso Ebola e uno sferoidale con protuberanze coronavirus (o nel caso dei batteri tra forme bastoncellari e cocchi, Gram positivi e negativi, ...) ma non possiamo sfruttare appieno il concetto di specie come faremmo con gli animali sia per ragioni di riproduzione asessuata che per la capacità di questi di scambiare materiale genetico anche tra membri molti diversi tra loro (problema questo alla base della diffusione della resistenza agli antibiotici).
Qui il molecolare non è più solo uno strumento ma LO strumento, usato ad esempio per studiare la parentela tra il SARS-CoV-2 e il primo membro "famoso" dei coronavirus il SARS-CoV (l'agente causale della SARS). Grazie a questo approccio si è compreso che il primo non è una variante del secondo ma un ramo laterale così come le scimmie attuali non sono i nostri antenati ma "cugini" con cui condividiamo un antenato comune. Solo semantica? No, comprensione della dinamica evolutiva.
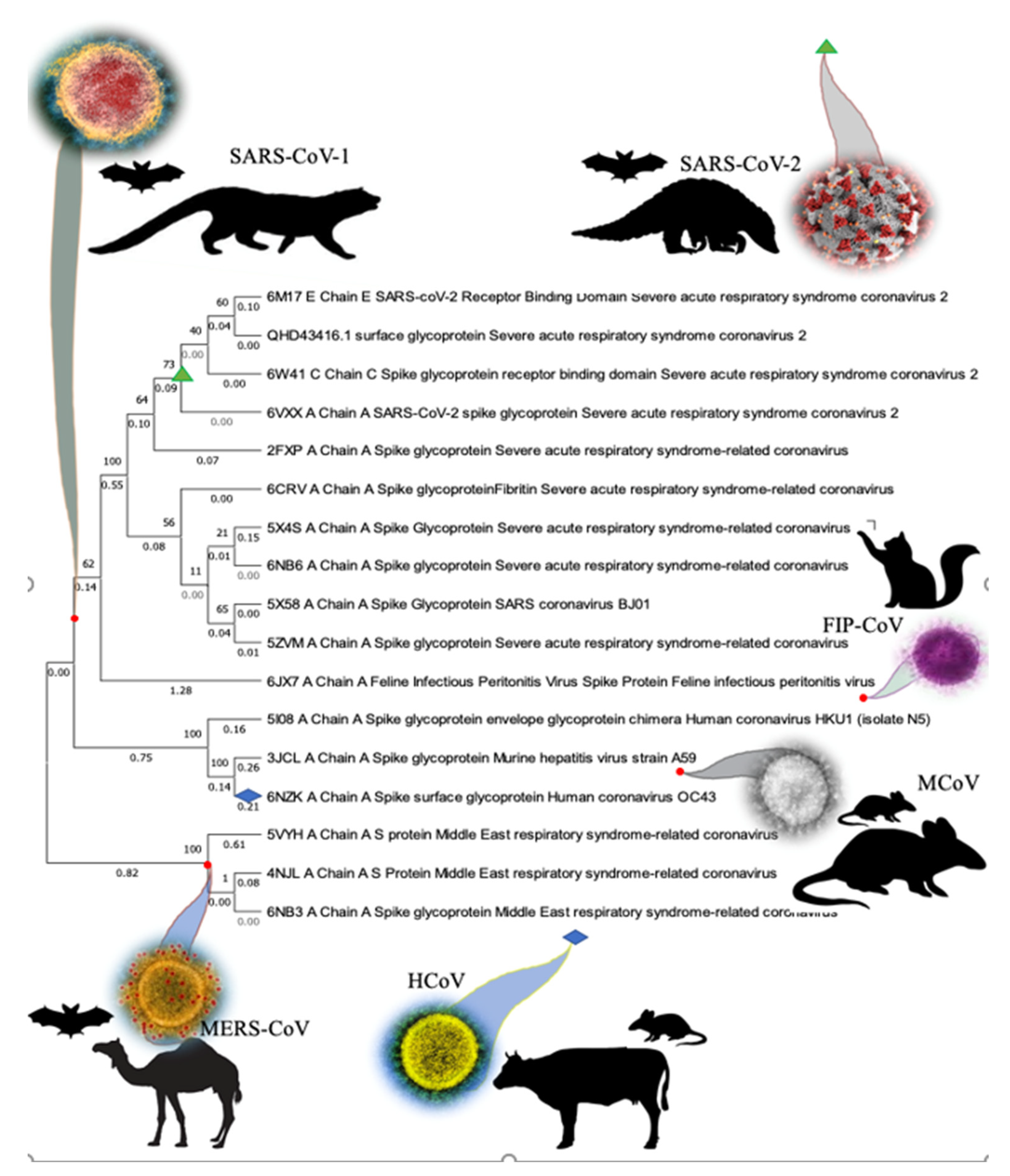 |
| Albero filogenetico ottenuto mediante ricostruzione bioinformatica. Clicca per ingrandire o vai alla pagina dell'articolo originale (credit: Antibodies 2021, 10(1) 3 ) |
Nello studio dei virus molte sono le informazioni, molecolari e non, che contribuiscono alla caratterizzazione. Si inizia con la prima scrematura morfologica per passare al tipo di genoma posseduto (DNA, RNA, singolo o doppio filamento, numero di segmenti, ...) per cercare infine di rispondere a quesiti tipo: quanti e quali sono i geni codificati, e come sono organizzati nel genoma; com'è il ciclo riproduttivo; le caratteristiche delle proteine di superficie; etc.
Ed ecco allora che finalmente ci si imbatte nel termine ceppo (strain) che in verità non ha valenza analitica univoca ma dipende molto da chi lo usa o dal contesto in cui è usato. Termine in ogni caso più usato del ben più generico mutante, che può indicare sia un dato fenotipico che il portatore di una singola mutazione non silente ma funzionalmente non caratterizzante.
Il termine ceppo è ancora più elusivo da spiegare del termine razza animale (cani, bovini, etc) in quanto almeno in quest'ultimo caso si riferisce alla stessa specie con tratti morfologici univoci risultante da incroci selettivi. Con ceppo si intende un isolato emerso da colture di laboratorio come potrebbero essere le cellule di una singola colonia batterica presente in una piastra di Petri.
Alcune persone usano questo termine per definire una "versione" specifica del virus isolata in un determinato momento. Altri invece lo usano per indicare un particolare "gruppo" in cui ogni membro è più simile agli altri membri del gruppo che ai membri di altri gruppi.
Confusi? Ricordate che ogni volta che avviene una infezione produttiva in una cellula la progenie virale porterà molte varianti e se queste daranno origine a ceppi distinti sarà solo soltanto perché hanno avuto (maggiore) successo rispetto alle "sorelle" nel generare una linea propria.
Ci imbattiamo così nel termine genetic lineage (linea genetica) che meglio dà il senso di quello che si vuole comunicare; anche qui però si impone una contestualizzazione perché potrebbe indicare sia un (oramai) ramo principale dell'albero genetico che una sottolinea, un piccolo ramo all'interno di un ramo più grande. In ogni caso il termine ci informa che stiamo guardando una popolazione in evoluzione e che ci si trova di fronte alla domanda se quanto questa linea vale la pena di essere seguita o se è un evento spurio che verrà diluito in fretta nel mare magnum della progenie virale.
La linea NON è nulla di statico ma accumulerà altre mutazioni la cui rilevanza ne determinerà il successo o la scomparsa
Ricapitolando se sentite il termine "ceppo" non è la descrizione migliore perché dipende se si sta parlando del virus isolato da uno specifico campione / paziente o della sottofamiglia di isolati correlati a quello originale che ha dato origine all’epidemia. In genere tra gli addetti ai lavori lo si usa quando si intende riferirsi al secondo caso (il messaggio sotteso è la trasmissione del ceppo).
Il SARS-CoV-2 ci offre un perfetto, e attuale, esempio dello sforzo di classificare e tracciare l'eterogeneità del pool virale in circolo in linee e sotto-linee: lo scopo è monitorare la dinamica della diffusione del virus sia a livello locale che globale.
Data la palese eterogeneità, a cosa sono riferite le differenze? La sequenza "standard" deriva sia dal primo isolato virale che dalla definizione di una "sequenza di consenso" che riporta la base nucleotidica più frequentemente osservata in ogni posizione del genoma, ben sapendo che c'è una variazione minore in ogni posizione tra le zilioni di particelle virali generate durante un singolo ciclo infettivo a causa delle mutazioni. Nel caso in esame il riferimento è al virus isolato a Wuhan all'inizio dell'epidemia.
Il seguente grafico, tratto dall'articolo "A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology" rende l'idea di cosa intendevo con eterogeneità all'interno del virus causa del Covid19.
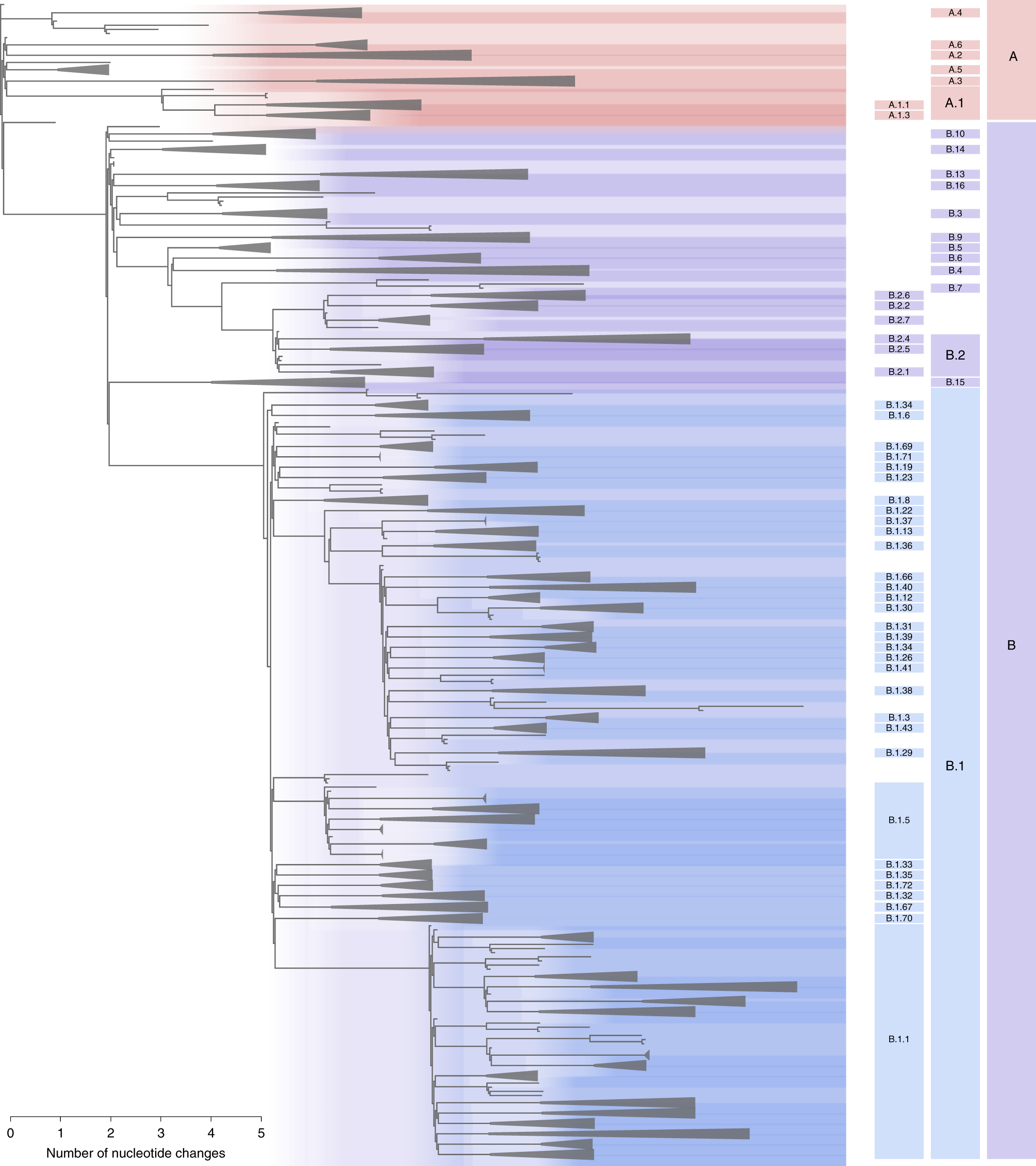 |
| Cliccate per ingrandire o andate direttamente alla pagina originale dell'articolo. Le 3 colonne laterali a destra rappresentano tre diversi livelli di raggruppamento/divisione a seconda di quanto si vuole entrare nel dettaglio. Notate come le "punte" di ogni ramo abbiano la forma di un triangolo allungato per dire"qui sono presenti n rametti che non disegneremo individualmente". (credit: A. Rambaut et al, Nature Microbiology (2020)5, pp1403–07) |
Volete qualcosa di più dettagliato? L'albero filogenetico interattivo presente sul sito nextrain.org fa per voi. Qui compare un nuovo termine, CLADE, che in virologia fa da contenitore per i gruppi simili unicamente sulla base delle sequenze genetiche. Uno strumento fondamentale per seguire l'evoluzione di una linea senza confondersi con "mutazioni convergenti".
 |
| Cliccate per ingrandire o andate direttamente sul sito. |
Arrivare a questo livello di tracciamento è oggi possibile grazie all'evoluzione delle tecniche di sequenziamento (quando ai miei tempi sequenziare un gene era un lavoro di una equipe di ricercatori che durava anni) e ai software che permettono di analizzare e assemblare il genoma di migliaia di isolati virali in automatico che verranno poi assegnati a linee sulla base di una serie di "mutazioni che definiscono la linea”.
E il temine "mutazione"?
Le mutazioni sono qualsiasi cambiamento nel genoma dell'oggetto in studio; in teoria geni, ma in realtà anche regioni non codificanti e per esteso gli organismi portatori, siano essi multicellulari o microbi, una volta che tale alterazione sia stata fissata.
Le mutazioni sono eventi intrinseci e fondanti l'evoluzione, risultato sia di errori di copiatura durante la replicazione del genoma che (dove presente) errori compiuti dal sistema di riparazione del genoma su danni causati da agenti ambientali. La fase di copiatura è un passaggio critico tanto che le cellule (e alcuni virus) hanno evoluto meccanismi di proof-reading (correzione di bozze) per eliminare gli errori nel momento stesso in cui sono compiuti.
Molti virus privi di questo controllo, specialmente quelli a RNA il cui macchinario replicativo è di per sé a rischio errore, avranno un elevato tasso di errore (tra 10−6 e 10−4) che se da una parte comporta la produzione di virus non funzionali, dall'altra assicura una variabilità intrinseca che rende il virus in grado di rispondere prontamente alle sfide ambientali, tra cui eludere la risposta immunitaria.
Curiosamente il SARS-CoV-2 in particolare e i coronavirus in generale sono virus a basso tasso di errore (compatibilmente con l'essere virus a RNA) essendo dotati di proof-reading.
Ma parliamo pur sempre di virus per cui quando una cellula viene infettata, anche da un singolo virione, la popolazione virale che emergerà (dati i numeri) avrà una certa probabilità di avere varianti "utili": la probabilità è funzione sia della "lotteria" delle mutazioni (gene e tipo di mutazione) che di puro caso(il virus emerso troverà qualcuno da infettare?)
Quando un virus entra in contatto con un nuovo bersaglio, ogni anello della catena di trasmissione rappresenta un nuovo punto di partenza nell'evoluzione del virus sia questo un ramo minore di una linea preesistente o un nuovo ramo dell'albero come quello l'evento zoonotico che ha dato origine alla pandemia (un virus "nato" in un pipistrello che ha differenza dei suoi consimili aveva acquisito mutazioni tali da rendere permissiva l'infezione e la replicazione in cellule umane).
Ogni individuo infetto genera una nuvola di varianti e una singola variante può infettare l'individuo successivo. Il suo successo dipenderà dalla capacità di sfuggire agli anticorpi, efficienza di replicazione, virulenza, riconoscimento del bersaglio, …. . Della sola proteina Spike sono state descritte 4 mila mutazioni, ma solo poche sono "utili" al virus e ancora meno quelle emerse come parte costituenti una linea.
Questo fenomeno spiega per quale motivo le persone con infezione cronica o con durata durata molto maggiore del normale, come tipico dei pazienti nelle ICU sottoposti a trattamenti sia sintomatici che antivirali, sono i perfetti selettori di virus più efficaci data la forte pressione selettiva.
Riassumendo, variante è un termine “gergale” per indicare qualsiasi genoma virale là fuori che abbia acquisito mutazioni rispetto al virus originale. Un po' come il termine "mutante". Siamo tutti mutanti in un certo senso, tranne per il fatto che alcune delle nostre mutazioni sono più visibili esternamente di altre a causa dei loro effetti.
Noi stessi siamo mutanti rispetto allo zigote originale e alcune cellule più di altre. Senza scomodare la ben nota ricombinazione genomica che avviene durante la maturazione dei linfociti, cito i neuroni in cui si ha l'attivazione trasposonica (LINE-1 elements, che altro non sono che retrovirus vestigiali) durante lo sviluppo embrionale.
Da un punto di vista pratico e comunicativo l'unico termine utile è Variant Of Concern (VOC).
Il VOC definisce una famiglia di varianti caratterizzata da un insieme caratteristico di mutazioni che, per definizione, sono fonte di preoccupazione per loro caratteristiche come la capacità di "sfuggire" alla neutralizzazione da parte di anticorpi indotti da vaccini o infezioni precedenti, aumentata trasmissibilità, virulenza o resistenza agli antivirali.
Quando leggete sui media variante in realtà si intende sempre VOC. Ci sono innumerevoli varianti del Sar-CoV-2 ma solo un piccolo numero ha acquisito mutazioni da monitorare: VOC sudafricana, inglese, etc etc
La linea B.1.1.7 (alias variante inglese alias VOC-202012/01) è definita da mutazioni che non sono necessariamente specifiche per questa linea ma il cui insieme lo è. Nello specifico si tratta di 23 mutazioni (rispetto all'originale di Wuhan), 8 a carico della proteina Spike e di queste 3 delle quali con rilevante potenziale biologico (vantaggio selettivo)
***
Nota. Quanto scritto dovrebbe anche rendere evidente il problema nella
nomenclatura in uso per descrivere due appartenenti al genere Homo, i sapiens e i neanderthalensis.
Se infatti appartenessero a specie distinte, come la nomenclatura farebbe supporre (Homo sapiens e Homo neanderthalensis) non avrebbero, per
definizione, potuto generare progenie fertile di cui noi (tutti i
sapiens non africani) portiamo tracce nel genoma e quindi prova di
unioni fertile (vedi articoli precedenti "una unione non priva di problemi" e "Denisovani").
Molto più
verosimile che l'unione sia avvenuta tra proto-sapiens e proto-neanderthelensis in una fase in cui il processo di speciazione non si
era ancora completato, quindi sarebbe meglio parlare di sottospecie. Un
caso simile lo abbiamo tra scimpanzé e bonobo di cui si hanno tracce genetiche di accoppiamenti avvenuti in passato.

Nessun commento:
Posta un commento